#SourceCode Overview
The #SourceCode project allowed us to experience a real-world intersection between Biology and computer programming. In groups of four that consisted of a project manager, an artist, a biologist, and a bioinformaticist, our class compared the sequenced genome of an individual to the sequenced genomes of one thousand other individuals in order to understand their ethnic ancestry.
Cristina, The Bioinformaticist
As a bioinformaticist, my job was to write a computer code in Python that evaluates a single individual's sequenced genome and compares it to that of a thousand other people's genomes. These thousand individuals all have known ancestry, so we can determine the ancestry of our person by comparison. To be able to do this, I had to have basic background knowledge on Python, one of the most common computer programming languages. I completed the Python course on Codecademy to help me understand the language and more easily so I had a general grasp on what I was doing.
How did we get this genetic information?
We were able to get the information needed for this project by using 23_and_me.com and OpenSNP.org. 23_and_me is a company that takes samples of people’s cells and sequences their genomes. Some individuals chose to donate their genetic information to public scientific study, and can therefore upload their raw genetic file to a program called OpenSNP.org, which can be found online. Those who are looking for genetic information can access these donated files through OpenSNP.org.
def main():
parsed_23andme_genotypes = parse_23andme_file()
parse_1KG_file(parsed_23andme_genotypes)
def parse_23andme_file():
input_file = open("H:\example_23andme_data.txt", "r")
genotypes = {}
for line in input_file:
line = line.rstrip("\n")
if line[0] == "#":
print line
else:
x = line.split("\t")
genotypes[(x[1], x[2])] = x[3]
return genotypes
def parse_1KG_file(genotypes_for_1KG):
input_file = open("H:\example_1KG.txt", "r")
output_file = open("H:\Obi_Wan_File.txt", "w")
for line in input_file:
line = line.rstrip("\n")
if line[0] == "#" and line[1] == "#":
output_file.write(line + '\n')
elif line[0] == "#" and line[1] != "#":
output_file.write(line + '\t' + 'my_sample'+ '\n')
else:
fields = line.split("\t")
if (fields[0], fields[1]) in genotypes_for_1KG:
chromosome = fields[0]
postition = fields [1]
REF=genotype [3]
ALT= genotype[4]
genotype = genotypes_for_1KG[(chromosome , position)]
allele_1 = genotype[0]
allele_2 = genotype[1]
if allele_1 == REF and allele_2 == REF:
output_file.write(line + '\t' + '0|0'+ '\n')
elif allele_1 == REF and allele_2 == ALT:
output_file.write(line + '\t' + '0|1'+ '\n')
elif allele_1 == ALT and allele_2 == REF:
output_file.write(line + '\t' + '1|0'+ '\n')
elif allele_1 == ALT and allele_2 == ALT:
output_file.write(line + '\t' + '1|1'+ '\n')
if __name__ == '__main__':
main()
In the first part of the code, the computer calls the file 23_and_me from the computer, and reads through it. When opened, 23_and_me file prints information about the individual’s chromosome position, and genotype.
In the second part of the code, the computer opens the 1KG file, the file that contains genetic information about one thousand other people with known ancestries, and compares it to the 23_and_me file. To do this, the information in the 23_and_me file needs to match the format of the 1KG file. So, through this second part of the code, the genotypes of the 23_and_me are compared to the reference and alternate genotypes of the 1KG file. If the genotype matches the reference, a 0 is printed, but if it matches the alternate, a 1 is printed. Dr. Schultz, a local bioinformaticist, helped us organize the data into a graph using multidimensional scaling to display the ethnic background of our given sample( 23_and_me).
def main():
parsed_23andme_genotypes = parse_23andme_file()
parse_1KG_file(parsed_23andme_genotypes)
def parse_23andme_file():
input_file = open("H:\example_23andme_data.txt", "r")
genotypes = {}
for line in input_file:
line = line.rstrip("\n")
if line[0] == "#":
print line
else:
x = line.split("\t")
genotypes[(x[1], x[2])] = x[3]
return genotypes
def parse_1KG_file(genotypes_for_1KG):
input_file = open("H:\example_1KG.txt", "r")
output_file = open("H:\Obi_Wan_File.txt", "w")
for line in input_file:
line = line.rstrip("\n")
if line[0] == "#" and line[1] == "#":
output_file.write(line + '\n')
elif line[0] == "#" and line[1] != "#":
output_file.write(line + '\t' + 'my_sample'+ '\n')
else:
fields = line.split("\t")
if (fields[0], fields[1]) in genotypes_for_1KG:
chromosome = fields[0]
postition = fields [1]
REF=genotype [3]
ALT= genotype[4]
genotype = genotypes_for_1KG[(chromosome , position)]
allele_1 = genotype[0]
allele_2 = genotype[1]
if allele_1 == REF and allele_2 == REF:
output_file.write(line + '\t' + '0|0'+ '\n')
elif allele_1 == REF and allele_2 == ALT:
output_file.write(line + '\t' + '0|1'+ '\n')
elif allele_1 == ALT and allele_2 == REF:
output_file.write(line + '\t' + '1|0'+ '\n')
elif allele_1 == ALT and allele_2 == ALT:
output_file.write(line + '\t' + '1|1'+ '\n')
if __name__ == '__main__':
main()
In the first part of the code, the computer calls the file 23_and_me from the computer, and reads through it. When opened, 23_and_me file prints information about the individual’s chromosome position, and genotype.
In the second part of the code, the computer opens the 1KG file, the file that contains genetic information about one thousand other people with known ancestries, and compares it to the 23_and_me file. To do this, the information in the 23_and_me file needs to match the format of the 1KG file. So, through this second part of the code, the genotypes of the 23_and_me are compared to the reference and alternate genotypes of the 1KG file. If the genotype matches the reference, a 0 is printed, but if it matches the alternate, a 1 is printed. Dr. Schultz, a local bioinformaticist, helped us organize the data into a graph using multidimensional scaling to display the ethnic background of our given sample( 23_and_me).
Understanding the Data
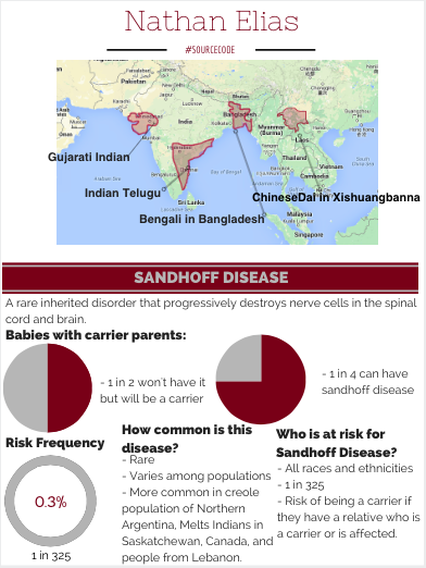
We used our data to make this inforgraphic that represents Nathan Elisas's ancestry. We also determined that he is susceptible to Sandhoff Disease based on his sequenced genome.
We also used our code to create a beautiful piece of art that represents our team.

This was not easy.
The most significant memory I will have from the #Sourcecode Project ten years from now will be that I pushed myself beyond my own expectations. When it comes to project work, I’m usually the most comfortable with being a project manager, and I can say that sometimes, that seems to be my comfort zone. I knew that I was not ready to jump into the role of being a bioinformaticist- there are many other people who are more experienced with computers and technology than I am, who I was certain were going to be the bioinformaticists. But, I took a risk by doing something that I thought I would never be able to do by deciding to work on the code that compared one person’s sequenced genome to a thousand other people’s to understand their genetic ancestry. I felt really challenged during this project, and when other students seemed like they had much better understandings of the code than I did (even if they had the same amount of experience with programming), I felt like giving up. Caro and I worked tirelessly, asking questions whenever we could, and working hard to understand the concepts that we needed to successfully write the code. When I realized that Mackenzie wasn’t our only source for help, I started to meet with Lynnee constantly during project work time- she is a fountain of knowledge, and she did an excellent job of explaining her understanding of the code. Even as I worked with Lynnee, I would often be lost and confused about what we were trying to do, and I had to become comfortable with not knowing the answer- and being brave enough to search for it. Looking back on this part of the project, I realize that I learned a lot about working with my peers and learning from them. I learned that I can’t be afraid to ask questions when I don’t understand something, and to find resources in all my surroundings. Once I started getting a better grip on the code, I often worked with Gabe to pass along what I knew. This experience was valuable, because when I started helping him, I realized how much I really didn’t know what I was doing. I had to go back to work with people like Lynnee and Mackenzie to ask questions about the work that I had already done. This process of learning and understanding seemed endless during the sourcecode project, but the work paid off. By the night of exhibition, I felt confident in my ability to explain what my code did and how it worked.
This project taught me much more than how to write a challenging code using Python. I learned how to find and use resources to educate myself and complete a difficult task, developed my resilience and problem-solving skills, and expanded my peer-education skills. However, the most important thing I learned was how to put aside my fear of failure in order to give myself opportunities to have new exciting experiences.
This project taught me much more than how to write a challenging code using Python. I learned how to find and use resources to educate myself and complete a difficult task, developed my resilience and problem-solving skills, and expanded my peer-education skills. However, the most important thing I learned was how to put aside my fear of failure in order to give myself opportunities to have new exciting experiences.
